Simptomi i znakovi ARG1-D često oponašaju one drugih neuroloških i neurometaboličkih poremećaja, kao što su drugi poremećaji ciklusa ureje (UCD), cerebralna paraliza (CP) ili nasljedna spastična paraplegija (HSP) 5,6.
Diferencijalna dijagnoza ARG1-D uključuje prepoznavanje kliničkih manifestacija bolesti udruženih
s visokim koncentracijama arginina u plazmi4-7.
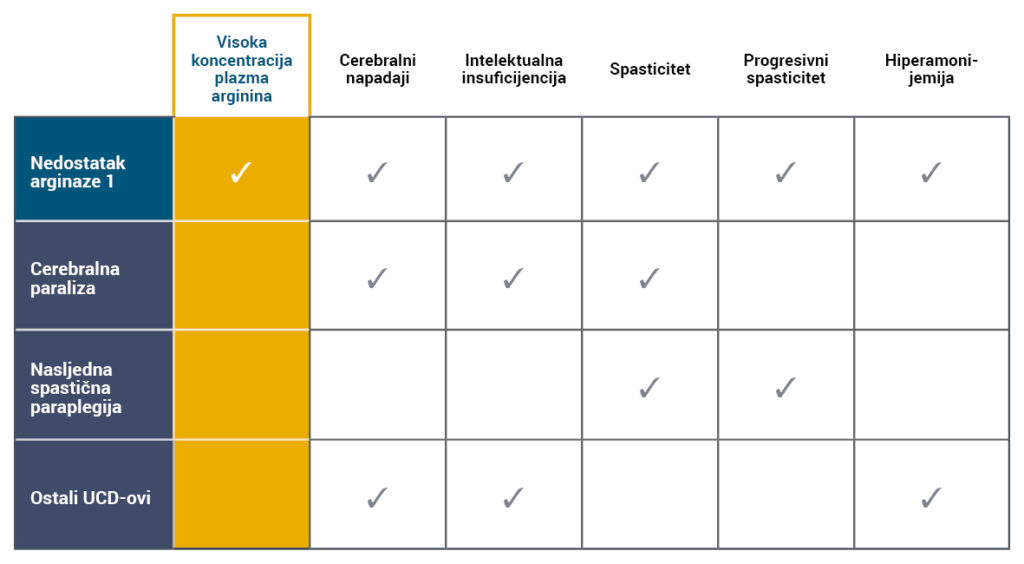
- Hiperamonemija nije karakteristično obilježje ARG1-D i akutne epizode hiperamonemije javljaju se rijetko4,8.
Zbog dijagnostičke ograničenosti novorođenačkog probira, ARG1-D se može propustiti iz brojnih razloga:
- Određivanje graničnih koncentracija arginina u probiru je problematično jer prijenos metabolita, poput arginina, s majke na dijete može ugroziti dijagnostičku pouzdanost rezultata9,10.
- Algoritmi probira i granične koncentracije arginina variraju9.
- ARG1-D nije uključen u panele novorođenačkog probira u većini europskih zemalj11.
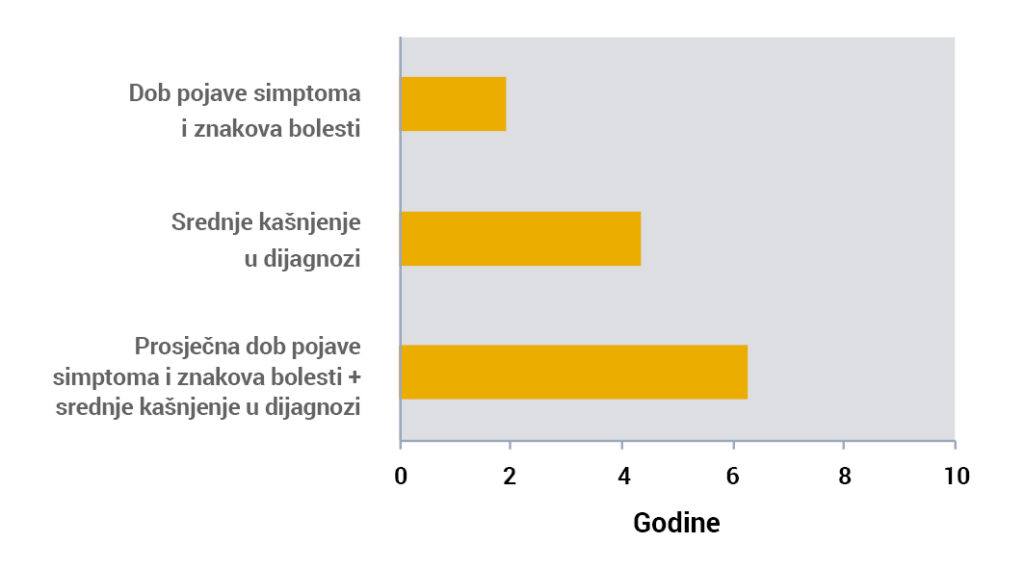
Kašnjenja u dijagnozi zajedno s kasnom pojavom simptoma dovode do početne intervencije tek u dobi od ~6 godina1.
Rutinsko mjerenje aminokiselina u plazmi praćeno analizom gena može potvrditi ARG1-D12,13.
Prije započinjanja dijagnostičkog postupka, važno je procijeniti kompletnu medicinsku, prehrambenu, obiteljsku i socijalnu anamnezu i obaviti temeljit fizikalni pregled.
Potvrdite postojanje visokih koncentracija arginina, koji uzrokuju manifestacije ARG1-D, rutinskim testiranjem3,12,13.


Ako je koncentracija arginina u plazmi visoka, analiza gena† može potvrditi dijagnozu .
†Zbog genetske heterogenosti genotipova ARG1, nisu identificirane sve mutacije koje uzrokuju ARG1-D.
Reference:
1. Huemer M, et al. J Inherit Metab Dis. 2016;39:331-340. 2. Edwards RL, et al. J Inherit Metab Dis. 2009;32:S197-S200. 3. De Deyn PP, et al. Hyperargininemia: a treatable inborn error of metabolism. In: Guanidino Compounds in Biology and Medicine. London, UK: John Libbey Company Ltd; 1997:53-69. 4. Burrage LC, et al. Hum Mol Genet. 2015;24:6417-6427. 5. Carvalho DR, et al. Pediatr Neurol. 2012;46:369-374. 6. Prasad A, et al. J Child Neurol. 1997;12:301-309. 7. Crombez EA, Cederbaum SD. Mol Genet Metab. 2005;84:243-251. 8. Scaglia F, Lee B. Am J Med Genet C Semin Med Genet. 2006;142C:113-120. 9. Therrell BL, et al. Mol Genet Metab. 2017;121:308–313. 10. Pitt JJ. Clin Biochem Rev. 2010;31:57-68. 11. Loeber JG, Platis D, Zetterström RH et al. Int J Neonatal Screening. 2021;7:15. 12. Sun A, et al. Arginase deficiency. In: Adam MP, et al, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2020. 13. Ah Mew N, et al. Urea Cycle Disorders Overview. 2003. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1217/. Accessed November 26, 2021. 14. Cai X, et al. Medicine (Baltimore). 2018;97:e9880. 15. Bélanger SA, et al. Paediatr Child Health. 2018;23:403-410. 16. Lüneburg N, et al. J Nutr. 2011;141:2186-2190.